
Dr. Douglas Kwazneski was helping a Pittsburgh surgeon remove an appendix when something jarring happened. The surgical stapler meant to cut and seal the tissue around the appendix locked up.
Kwazneski later turned to the Food and Drug Administration’s public database that tracks medical device failures and “there was nothing,” he said. Yet when he surveyed leading surgeons on the matter, he discovered that more than two-thirds had experienced a stapler malfunction, or knew a peer who did. Such failures can have deadly consequences.
Kwazneski had no idea the FDA had quietly granted the makers of surgical staplers a special “exemption” allowing them to file reports of malfunctions in a database hidden from doctors and from public view.
“I don’t want to sound overdramatic here, but it seemed like a cover-up,” said Kwazneski, who practiced in Pasco County, Fla., from 2016 through earlier this year.

After Dr. Douglas Kwazneski witnessed a surgical stapler malfunction, he surveyed leading surgeons and discovered that more than two-thirds had experienced a stapler malfunction, or knew a peer who did.
The FDA has built and expanded a vast and hidden repository of reports on device-related injuries and malfunctions, a Kaiser Health News investigation shows. Since 2016, at least 1.1 million incidents have flowed into the internal “alternative summary reporting” repository, instead of being described individually in the widely scrutinized public database known as MAUDE, which medical experts trust to identify problems that could put patients in jeopardy.
Deaths must still be reported in MAUDE. But the hidden database has included serious injury and malfunction reports for about 100 medical devices, according to the FDA, many implanted in patients or used in countless surgeries. They have included surgical staplers, balloon pumps snaked into vessels to improve circulation and mechanical breathing machines.
An FDA official said that the program is for issues that are “well-known and well-documented with the FDA” and that it was reformed in 2017 as a new voluntary summary reporting program was put in place for up to 5,600 devices.
Yet the program, in all its iterations, has been so obscure that it is unknown to many of the doctors and engineers dedicated to improving device safety. Even a former FDA commissioner said he knew nothing of the program.
KHN pored over reams of public records for oblique references to reporting exemptions. After months of questions to the FDA, the agency confirmed the existence of reporting-exemption programs and thousands of never-before-acknowledged instances of malfunctions or harm.
Amid the blackout in information about device risks, patients have been injured, hundreds of times in some cases, lawsuits and FDA records show.
“The public has a right to know about this,” said Dr. S. Lori Brown, a former FDA official who accessed the data for her research. She said doctors relying just on the public reports — and unaware that many incidents may be omitted — can easily reach the wrong conclusion about the safety record of a particular device.
The FDA has also opened additional — and equally obscure — pathways for device makers to report thousands of injuries brought to light by lawsuits or even deaths that appear in private registries that medical societies use to track patients. Those exemptions apply to risky and controversial products, including pelvic mesh and devices implanted in the heart.
FDA spokeswoman Deborah Kotz confirmed that the “registry exemption” was created without any public notice or regulations. “Any device manufacturer can request an exemption from its reporting requirements,” she said in an email.
I don’t want to sound overdramatic here, but it seemed like a cover-up.
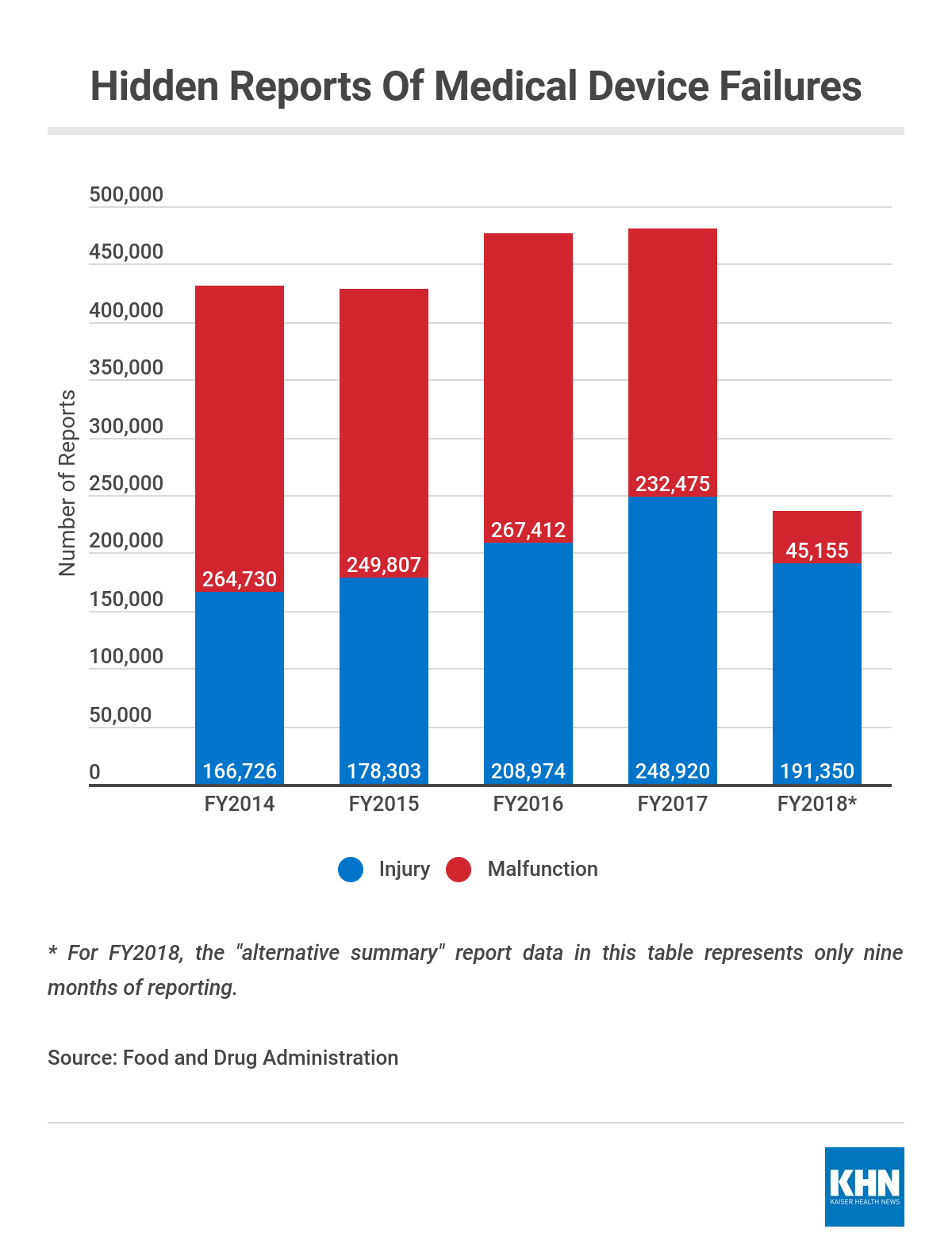
Agency records provided to KHN show that more than 480,000 injuries or malfunctions were reported through the alternative summary reporting program in 2017 alone.
Alison Hunt, another FDA spokeswoman, said the majority of device makers’ “exemptions” were revoked that year as a program took shape that requires a summary report to be filed publicly.
More than a million reports of malfunctions or harm spanning about 15 years remain in a database accessible only to the FDA. But with the agency’s new transparency push, the public may find a public report and submit a Freedom of Information Act request to get information about incidents. A response can take up to two years.
The long-standing exemption program “has allowed the FDA to more efficiently review adverse events … and take action when warranted without sacrificing the quality of our review or the information we receive,” Hunt said in an email.

Madris Tomes
To those outside the agency, though, the exceptions to the reporting rules are troubling. They strike Madris Tomes, a former FDA manager, as the agency surrendering some of the strongest oversight and transparency powers it wields.
“The FDA is basically giving away its authority over device manufacturers,” said Tomes, who now runs Device Events, a website that makes FDA device data user-friendly. “If they’ve given that up, they’ve handed over their ability to oversee the safety and effectiveness of these devices.”
Doctors, like Kwazneski, who have turned to the public data to gauge the risks of surgical staplers have seen little. He wrote about the “unacknowledged” problem of stapler malfunctions in a 2013 article in the journal Surgical Endoscopy. In 2016, while reports of 84 stapler injuries or malfunctions were openly submitted, nearly 10,000 malfunction reports were included in the hidden database, according to the FDA.
Device maker Medtronic, which owns stapler maker Covidien, has been described as the market leader in surgical staplers. A company spokesman said that the firm has used reporting exemptions to file stapler-related reports through July 2017. Ethicon, the other major stapler maker, said it has not.
The public database shows that Medtronic has reported more than 250 deaths related to staplers or staples since 2001.
Mark Levering, 62, nearly lost his life after a stapler malfunction early last year, according to a lawsuit filed by his family. His surgeon has testified that a surgical stapler misfired during his liver surgery at ProMedica Toledo Hospital in Ohio.
Staff performed CPR for 22 minutes while surgeons rushed to suture the severed vein. He emerged from a coma unable to walk or consistently recognize his wife and son. The surgeon, hospital and device maker Covidien have denied allegations of wrongdoing in an ongoing legal case.
Told of the reporting “exemption” for surgical staplers, his wife, Doris Levering, was incredulous.
“Why would this information not be made available to doctors? The true information — I mean the actual numbers …” she said. “People’s lives are at stake. Mark’s life will never be the same.”

Doris Levering says her husband, Mark, underwent a procedure to remove an abscess from his liver and ended up in a coma for weeks.
The Stapler Problem
The sheer number of malfunctions made surgical staplers an easy pick for the new alternative summary reporting program at its inception nearly 20 years ago, according to Larry Kessler, a former FDA official and now a University of Washington health services professor.
Surgical staplers have a unique ability to help — or harm — patients. The device is designed to cut and seal tissues or vessels quickly, often during minimally invasive surgeries. When it fails to seal a major blood vessel, medical staff can quickly shift into “code blue” mode to rescue a patient from bleeding to death.
The severity of some of the injuries caught former FDA official Brown’s attention in the early years of its reporting exemption. Her 2004 article on stapler mishaps, published in the Journal of the American College of Surgeons, accounts for one of the few places in public records where an FDA authority mentions the “alternative summary” program. She found that in the first 28 months of filing to the hidden database, stapler makers filed more than 5,100 reports of malfunctions or injuries.
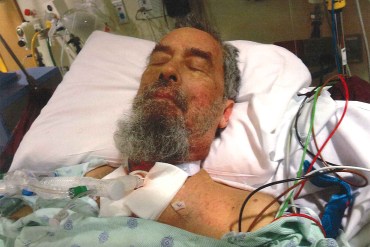
Mark Levering, now 62, was in a medically induced coma and had several surgeries after a surgical stapler malfunctioned during a Feb. 17, 2018, liver surgery. His wife, Doris Levering, said in a deposition that he did not open his eyes after the initial surgery until March 13.
She also noted that the publicly reported 112 stapler-related deaths in patients aged 22 to 91 from 1994 to 2001 were a “reason for concern.”
In the public data filed since, it would appear that the staplers rarely misfire. In 2011, only 18 injury or malfunction reports were filed publicly. Last year, the number was 79.
Lawsuits detail how quickly a stapler failure can turn a smooth surgery into a catastrophe.
In Michigan, Eugene Snook’s surgeon was in the process of removing part of his lung when he cut but couldn’t seal a major vessel due to a “stapler malfunction,” the surgeon said in sworn court testimony. Snook, then 59, had no detectable blood pressure for four minutes during the 2012 surgery.
The damage to Snook’s artery was so great, his surgeon decided to remove his lung completely, medical records filed in court say. Snook sued stapler maker Covidien, which in court records said there was no proof the stapler was unsafe when it left Covidien’s control and also that the surgeon used it improperly. The case reached a confidential settlement in 2017.
Another surgeon attempting to remove a benign liver growth from April Strange, 33, in 2013, testified that a stapler malfunction caused the woman to bleed to death. Strange, of central Illinois, left behind a husband and two daughters, then 6 and 8.
The stapler was thrown out after surgery, court records say. Covidien argued in court records that Ryan Strange couldn’t prove that the stapler had a specific defect.
Covidien reached an agreement to settle the family’s claims for $250,000, part of a larger settlement in the case.
Doctors initially thought Mark Levering had liver cancer. So when the diagnosis came back as an abscess that needed to be surgically removed last February, it came as a relief to his wife, Doris.
That relief turned to dread the day of surgery. The procedure was supposed to last two hours, she said. But the surgery hit a snag when the stapler “misfired,” according to the surgeon, causing so much bleeding that the minimally invasive procedure was converted to an open procedure so the doctor could suture the vein.
Levering underwent CPR for 22 minutes. A code blue was called, a nurse testified. Levering lost 3 quarts of blood — about half the blood in his body. He was put on life support and would remain in a coma for weeks.
After Levering reopened his eyes, it was clear that the man who used to tend to stray cats and enjoy dinner out with his family was gone. Levering could no longer walk, comb his hair or recognize the letters of the alphabet.
Doris and Mark Levering have sued the doctor, hospital and surgical stapler maker, alleging that the device caused Mark’s bleeding and brain injury. The surgeon has acknowledged in sworn testimony that the stapler malfunctioned, but denied other wrongdoing. The hospital said in a legal filing that its actions were “prudent, proper” and “lawful.”
Covidien denies any defect with the stapler or that it caused Levering’s injuries. A spokesman for parent company Medtronic declined to comment further on any lawsuit but said that “we always make patient safety our top priority” and that the company complies with FDA requirements.
The company’s reports of stapler problems in the public database remain relatively low. But in 2018, with the reporting exemption gone, a spike of reports emerged for Covidien’s staples — not to be confused with staplers. While Medtronic reported 1,000 staple malfunctions or injuries in 2015, the number soared to 11,000 for 2018.
Rolling Out The Program
The alternative summary reporting program started two decades ago with a simple goal: to cut down on redundant paperwork, according to officials who were at the FDA at the time.
Kessler, the former FDA official, said the program took shape after scandals over under-reporting of device problems spurred changes allowing criminal penalties against device companies.
Soon, thousands of injury and malfunction reports poured into the agency each month, with about 15 staff members dedicated to reviewing them, Kessler said. Many reports were so similar that reviewing them individually was “mind-numbing.” Kessler went to the FDA’s legal department and to device manufacturers to propose a solution.
Device makers would be able to seek a special “exemption” to avoid reporting certain complications to the public database. The manufacturers would instead send the FDA a spreadsheet of injury or malfunctions each quarter, half-year or year.
That way, Kessler said, reviewers could quickly look for new problems or spikes in known issues. When the program launched in 2000, the list of exempted devices was made public and only a few devices were involved, Kessler said.
“I don’t know why it’s not [made public] now,” he said. “I’m surprised about that.”
Starting in September, KHN filed Freedom of Information Act requests for “exemption” agreements and reports for several medical devices. Health and Human Services officials denied an appeal to provide some of the records quickly, concluding there was no “compelling need” for haste. For one request, the records were estimated to arrive in 22 months.
The FDA did provide some top-level data. It shows that from 2014 through 2017, the overall number of alternative summary reports filed by device makers rose from 431,000 to 481,000.
The FDA declined to provide a complete list of “about 100” devices that have been granted reporting exemptions over the years, but confirmed that exemptions have been used for mechanical breathing machines and balloon pumps, known as intra-aortic balloon pumps, inserted in the vessels of people with circulation problems.
An FDA spokeswoman said “alternative summary” exemptions remain in place for pacemaker electrodes and implantable defibrillators.
Matthew Baretich, a biomedical engineer in the Denver area, said he helps several area health systems analyze device-related patient injuries and make equipment-purchasing decisions.
He said he regularly scans the FDA’s public device-injury reports. Asked about “alternative summary” reports, he said, “I’ve got to tell you, that’s a new term to me.”
Bruce Barkalow, president of a Michigan-based biomedical engineering firm, said he’s the guy government officials, attorneys or device makers call if someone gets a pacemaker and dies in the shower three days later.
In an interview, he said he was not aware of the reports, either. He said they may appear to the FDA to be a “nothing burger,” but the data would be meaningful to his forensic investigations.
The ECRI Institute, a nonprofit leader in medical device safety, declined to provide an engineer for an interview. Educating hospital leaders and health providers, the institute issues an annual “Top 10” in medical technology hazards. Its tagline: “Separating fact from fiction in healthcare.”
Among the institute’s “top medical device subject matter experts,” spokeswoman Laurie Menyo said in an email, “none of them had any familiarity with FDA’s Alternative Summary Reporting Program.”
Even Dr. Robert Califf, former FDA deputy commissioner and commissioner from 2015 to 2017, said in an interview that he was unaware of the program. “Never heard anything about it,” he said. “It’s interesting.”
Companies that get the exemptions tend to be very “tight-lipped” about them, said Christine Posin, a former device firm manager and consultant to device companies.
The relative secrecy around the program can give them an advantage, she said. For instance, sales representatives can print out only the public reports of device problems, ignoring what’s buried elsewhere.
That creates a business opportunity to persuade a doctor to try a different device. “‘We have a good product that does the same thing,’” Posin said a sales representative might tell a physician.
Exemptions Multiply
The FDA has spent millions, convened experts and pledged to improve its work in device safety in recent years. All the while, it has quietly opened new avenues for the makers of controversial and risky devices to file injury and even death reports with little public review.
Pelvic mesh is one example. The fabric-like device has long been used to hold up pelvic organs in women experiencing organ prolapse. In 2011, the FDA issued a “safety communication” saying “serious complications” like pain or infection were “not rare.”
The agency soon reclassified the device, ordered safety studies and saw most mesh makers remove the device from the market.
Behind closed doors, though, the agency has since granted pelvic mesh makers a special exemption from reporting injuries to the public, according to the FDA and mesh makers who were asked about the practices.
Under what the FDA calls the “litigation complaint summary reporting” exemption, device makers can file a single “injury” report. Attached to the summary report, device makers have sent the FDA a spreadsheet with as many as 1,175 reports of patient injuries, based on allegations in lawsuits.
To someone tallying the overall number of injuries related to pelvic mesh, the report would appear as a single injury. It would take a sharp eye to find the summary report and a special request — taking up to two years to be filled — to get the details on the 1,175 cases submitted directly to the FDA.
According to the FDA, in 2017 alone, eight mesh makers used their exemptions to send nearly 12,000 injury reports to the FDA.
The FDA is basically giving away its authority over device manufacturers. If they’ve given that up, they’ve handed over their ability to oversee the safety and effectiveness of these devices.
Dr. M. Tom Margolis, a urogynecologist in the San Francisco Bay Area and an expert medical witness for those who are suing mesh makers, said a program that might hinder doctors relying on open FDA data to assess the risks of mesh is “horrible” and “unethical.”
“We need to know the good and the bad,” said Margolis, who treats patients in his urogynecology practice. “If you’re trying to hide complications from me, well that’s … wrong, my God, it’s heinous.”
The FDA issued the same kind of exemption to the makers of da Vinci surgical robots months after Johns Hopkins University School of Medicine researchers pointed out that the company was filing a notably small number of injury reports in the public database. Johns Hopkins professor Dr. Marty Makary noted in 2013 that the handful of reports sent to the FDA at the time were signs of a “haphazard” system that is “not independent and not transparent.”
Within months, the FDA allowed the makers of the robots to file a single report, noting that a spreadsheet sent straight to the FDA summarizes about 1,400 injuries alleged in lawsuits, with some injuries dating to 2004. Since then, the device maker has reported smaller batches of 99 and 130 injuries at a time.

Homa Alemzadeh
“This is very frustrating,” said Homa Alemzadeh, an assistant professor of computer engineering at the University of Virginia who is working with MAUDE data to create software to identify errors in real time or before they happen in surgeries performed by robots. She said she was not aware of the reporting exemption.
Under another reporting exemption, the FDA is allowing device makers to report hundreds of death cases in spreadsheets sent directly to the agency.
Under the “registry exemption,” device makers can summarize what they learn from registries that tend to be held by specialty medical societies, and track the use of a certain kind of device, according to FDA spokeswoman Kotz.
Kotz said the data in registries often falls short of the level of detail that the FDA seeks for the more thorough death reports that device makers are required to file.
Device makers filing such reports include Edwards Lifesciences, which makes the Sapien 3 valve that’s snaked through a vessel and implanted in the heart. Some hail the device as a breakthrough for saving patients from the trauma of open-heart surgery to replace a valve. Others raise concerns over limited data showing how long the valve will last in the body.
The summary reports offer potential patients few answers. Such reports document as many as 297 deaths or 1,800 injuries in a single filing, with virtually no detail readily available to the public. In all, Edwards has filed more than 1,800 Sapien 3 valve patient deaths as summaries since 2016.
Edwards spokeswoman Sarah Huoh said in an email that the FDA mandated the tracking of every patient who has the valve in the registry to provide “comprehensive evidence for device safety.”
“The approval of alternative reporting protects against duplicate reports coming from multiple sources,” Huoh said.
Another device, the MitraClip, is used to attach two flaps in the heart that are allowing blood to flow backward. The device has been controversial, with some scientists saying it is crucial for a certain subset of patients, and others pointing to the harm it can cause to the heart.
The FDA has allowed Abbott Vascular, which makes the MitraClip, to report as many as 347 deaths or 1,000 injuries in a single filing, also shipping the details straight to the agency, FDA records show.
An Abbott spokesman said in an email that the company has done clinical trials with thousands of patients to establish the MitraClip’s safety. He said the exemption was granted because data in the registry was stripped of patient identifiers, making it hard to know whether the company would be filing redundant reports to the FDA.
We need to know the good and the bad. If you’re trying to hide complications from me, well that’s … wrong, my God, it’s heinous.
Last year, the FDA finalized regulations for yet another summary reporting program. Under the newest system, device makers do not have to seek an exemption or notify the FDA that they’ll be filing a public summary report in MAUDE.
The FDA has deemed the makers of more than 5,600 types of devices eligible to file “voluntary malfunction summary reports.” Among them are some of the riskiest devices the agency oversees, including cardiac stents, leadless pacemakers and mechanical heart valves.
The growing cadre of exceptions to the injury- and death-reporting rules strikes Dr. Michael Carome, director of the Public Citizen Health Research Group, as a retreat by the FDA from making crucial information available for researchers and patients.
“It’s just another example of a flawed oversight system,” he said, “bent toward making it easier for industry rather than making protection of public health the primary goal.”
California Healthline reporter and producer Heidi de Marco contributed to this report.
[Correction: A previous version of this story mischaracterized FDA spokeswoman Alison Hunt’s description of the reports that go into a new FDA exemption program. It has been revised to eliminate the term “placeholder.”]
KHN’s “Hidden Harm” series on the FDA’s secret database of malfunctioning medical devices was honored on Oct. 2, 2019, with the prestigious Barlett & Steele Award for Investigative Journalism. The series prompted the FDA to publish its entire hidden database online, revealing 5.7 million device-related injuries or malfunctions.